SP A5
Translating genomic information to therapeutic targets for neuroblastoma using systematic loss-of-function screening
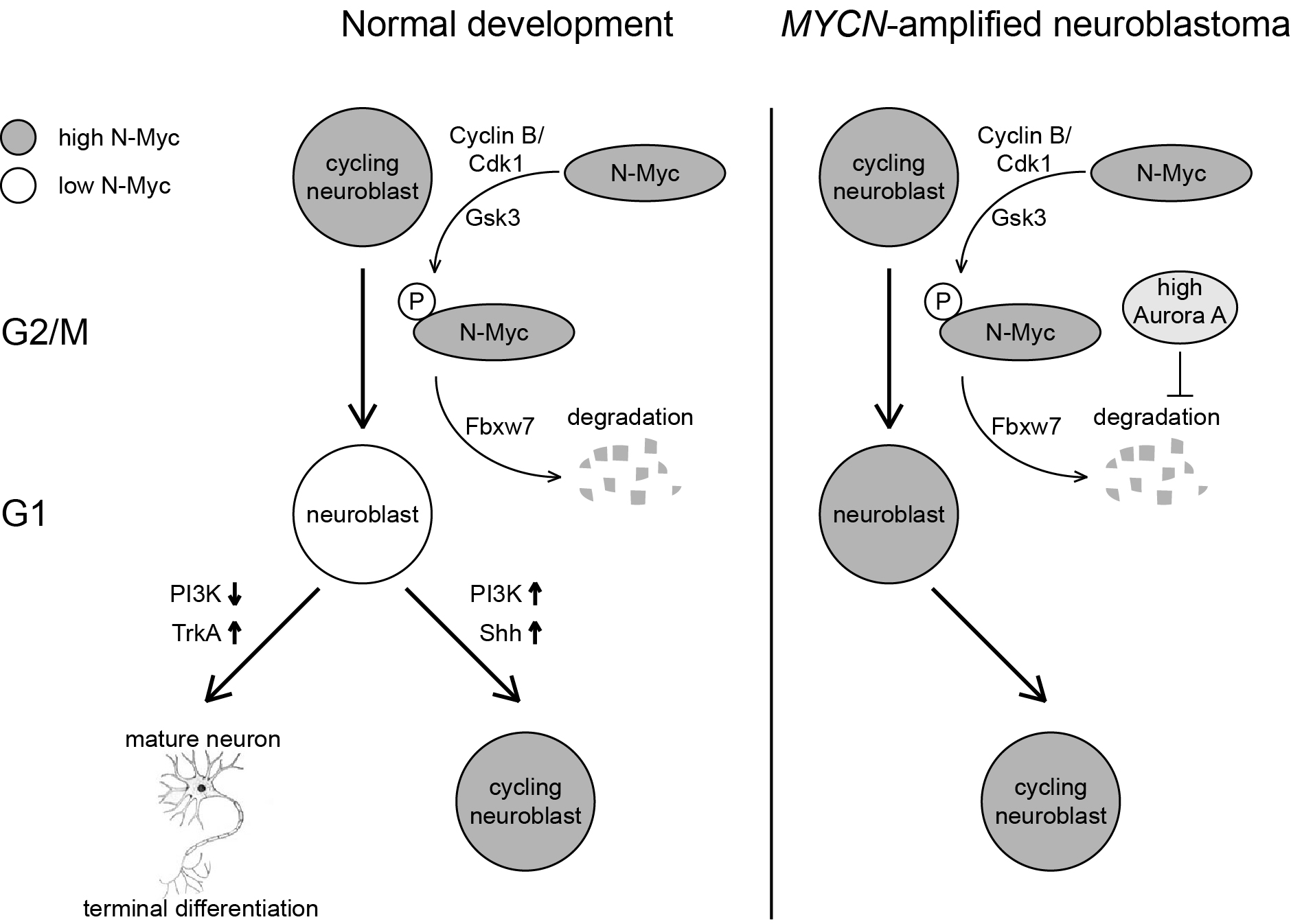
About 20% of human neuroblastomas harbor MYCN amplifications, resulting in high-level expression of an oncoprotein that promotes proliferation and self-renewal while blocking terminal differentiation of neuronal progenitor cells. Strikingly, neuroblastomas lacking MYCN amplifications harbor a diverse group of mutations, each capable of suppressing neurite outgrowth. Neurite outgrowth is indicative of terminal differentiation, suggesting that suppression of neurite outgrowth is a critical step during neuroblastoma development. Consistent with this notion, several stimuli can induce neuroblastomas to undergo terminal differentiation, and one of them, retinoic acid, is used in some current therapy regimens to retard tumor development.
While individual factors and mutations are known that block neuritogenesis in neuroblastoma, little is known about the underlying biochemical networks mediating this blockade and whether they contain critical nodes that can be inhibited to promote terminal and irreversible differentiation. We have established lentiviral shRNA screens that in combination with next-generation sequencing allow genome-wide loss-of-function screening. In WPA5, we aim to systematically identify druggable genes required for suppressing neuritogenesis and evaluate their therapeutic efficacy in preclinical models of neuroblastoma.