


SP 2
Central Resource I: Genomics and Epigenomics
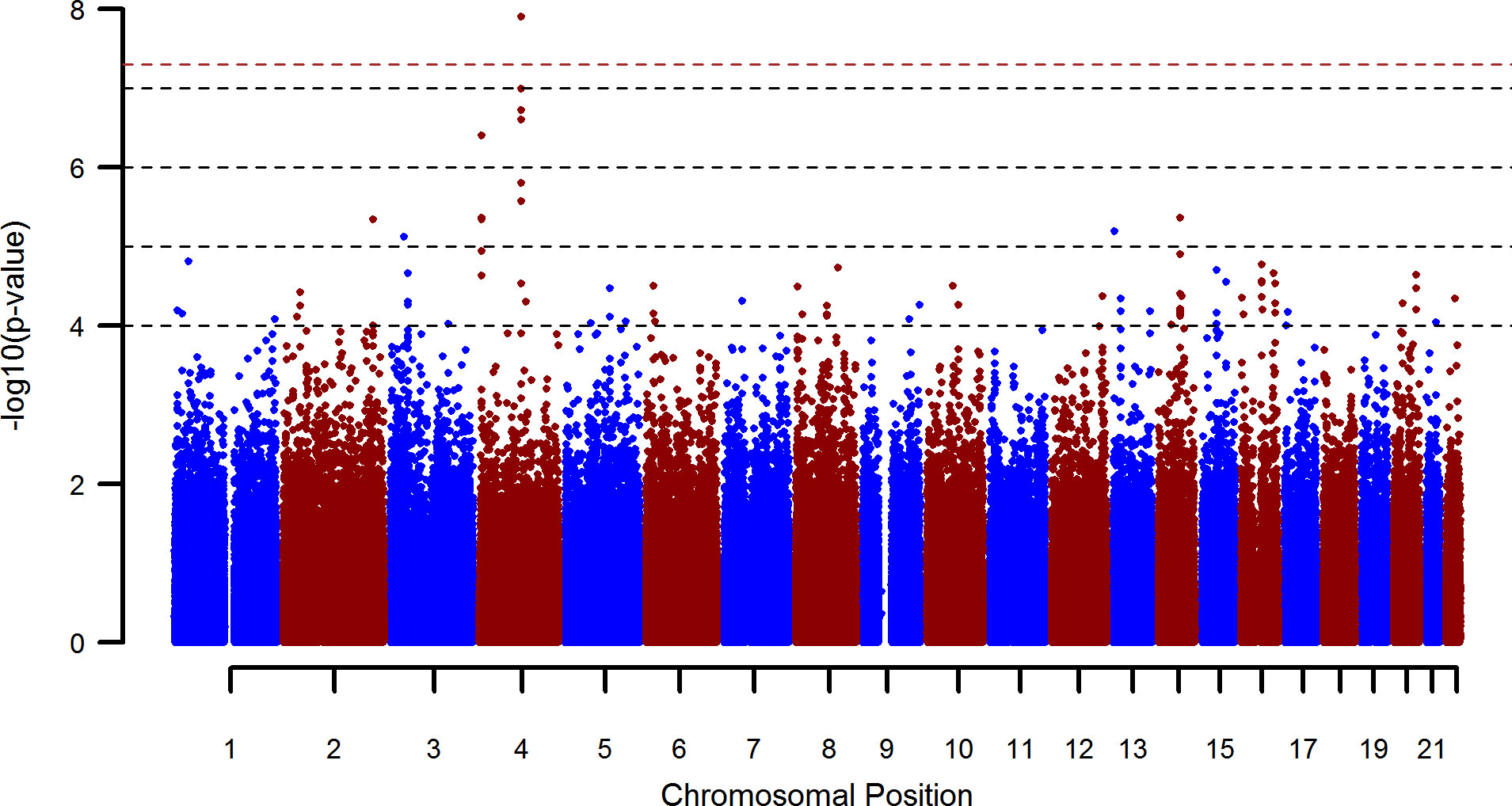
Alcohol dependence (AD) is a disorder with high prevalence, which leads to high social and economic costs. Formal genetic studies have shown that the heritability of AD is around 40–60%. In the past, funded by the National Genome Research Project, we have conducted candidate-, systematic genome-wide association- and pathway analyses, integrating also data from animal models. Using this approach, we were the first to generate genome-wide significant findings for AD, thus demonstrating the validity of a genetic approach in understanding the biological basis of the disorder (figure 1). These findings have since received independent support from other groups. Further investigation of one of the associated genes (GATA4) has demonstrated a correlation with relapse, as well as functional consequences in specific brain regions. Through subsequent multi-marker analysis, i.e. the simultaneous analysis of multiple genetic variants, we demonstrated that a large number of markers, each with a small effect, contribute to AD.
The aim of the central resource subproject 2 (SP2) is to identify genetic variants and epigenetic factors of relevance to AD etiology and disease-course. To achieve this, a range of approaches will be applied: (i) metaanalyses performed within international collaborations; (ii) pathway analyses to test for the joint contribution of functionally related markers with small effect sizes; (iii) resequencing of the most promising genes in order to identify rare variants with higher penetrance; (iv) identification of more homogenous patient subsets using imaging techniques for the assessment of neuronal patterns (endophenotypes); and (v) analysis of correlations between DNA-methylation and alcohol consumption patterns. We anticipate that the comprehensive analysis of genome wide association (GWA) and epigenomic data, and the integration of these data with findings generated within the other Consortium SPs, will implicate novel genetically- and epigenetically regulated genes in AD.