


TP1 - SASKit
Biomarker Discovery für Bauchspeicheldrüsenkrebs, Schlaganfall und deren Komorbidität basierend auf humanen, Tier und in-vitro Daten.
Die Bioinformatik konzentriert sich auf die Analyse der Transkriptomik- und Proteindaten sowie des Blutbilds und anderer klinischer/phänotypischer Daten zusammen mit öffentlichen Daten, um Biomarker zu identifizieren. Unser Ziel ist es, einfach zu interpretierende diagnostische/theranostische Biomarker für die Verschlechterung/Erholung und letztlich für therapeutische Maßnahmen zu finden. Das maschinelle Lernen von Biomarkern basiert zunächst auf einfachen korrelationsbasierten Ansätzen, die im Prinzip alle Messungen umfassen können, für die mehrere Zeitpunkte oder Bedingungen verfügbar sind. Die Interaktionen mit der höchsten Korrelation und die korrelationsbasierten Teilnetze werden im Sommer/Herbst 2023 im Detail untersucht. Zum Beispiel würden wir erwarten, dass die longitudinale Veränderung von entzündlichen Blutbestandteilen (z.B. CRP) mit der entzündungsbezogenen Genexpression über Gewebe, Altersgruppe und den gesamten Seneszenzstatus hinweg korreliert. Wir führen auch diverse Standard-omics-Analysen durch, wie z. B. Gene Ontology und Pathway Enrichment-Analysen. Sobald Endpunktdaten wie Progression/Überleben oder Genesungsdaten verfügbar sind, werden Cox-Hazard-Modelle, Support Vector Machines (SVM), Random Forests und neuronale Netze eingesetzt.
Eine wichtige Komponente unserer Analyse ist der Parallelogramm- und Transfer-Learning-Ansatz, bei dem wir die Expressionsdaten für eine Spezies/Gewebe-Kombination extrapolieren, sobald die Daten für drei andere Kombinationen bekannt sind. Wir haben inzwischen maschinelles Lernen für den Genexpressionsdatensatz von van der Velpen (2016) verwendet, um den Parallelogramm-Algorithmus zu optimieren. Neuronale Netze waren in der Lage, den Vorhersagefehler für das unbekannte menschliche Gewebe um 63 % zu reduzieren, verglichen mit der einfachen Annahme, dass die Expressionsverhältnisse im Gewebe und im Blut bei beiden Spezies genau gleich sind, und wir erzielten ähnlich gute Ergebnisse für öffentlich verfügbare Genexpressionsdatensätze, die zu unserem SASKit-Projekt passen (Maus und Mensch; Blut, Gehirn und Bauchspeicheldrüse, von den Portalen GTEx und MGI und von Srivastava (2020)). Wir konnten jedoch noch in keinem öffentlichen Datensatz gepaarte Proben für beide Arten finden. Darüber hinaus begannen wir mit einer eingehenden Untersuchung des Transfer-Lernens, die zu einer Übersichtsarbeit führte (Kowald et al., 2022). Insbesondere der "Transfer Variational Autoencoder" (trVAE) von Lotfollahi (2020) kann einen vierten Datensatz vorhersagen, der auf drei geeigneten Datensätzen basiert. Wir planen daher, trVAE zu verwenden, indem wir unsere Expressionsdaten von Mäusegehirn und -blut sowie von Patientenblut verwenden, um die Expression im menschlichen Gehirn zu extrapolieren.
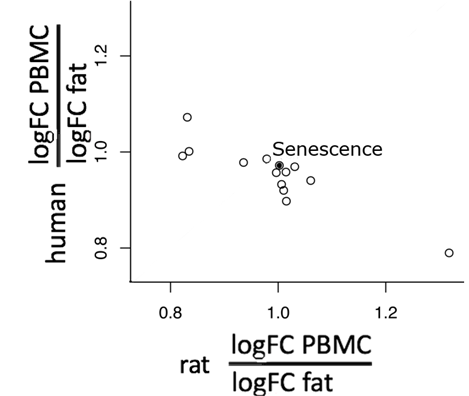
Parallelogramm Ansatz: Expressionsveränderungen für Transkripte, die an zellulärer Seneszenz und anderen Reaktionswegen beteiligt sind, werden aufaddiert und das Verhältnis Blut/Fett ist für Mensch und Ratte aufgetragen. Werte nahe 1 bedeuten eine große Ähnlichkeit zwischen Blut und Fettgewebe. In diesem einfachen Vorversuch liegt der Punkt für zelluläre Seneszenz (schwarz) nahe bei den Koordinaten 1/1, ebenso wie die Punkte diverser anderer Reaktionswege. Dies deutet auf ein gutes Ergebnis der Extrapolation hin.